















 40 / 48
40 / 48

40
|
Volume 2 Issue 2
S
upporting
Y
our
P
ractice
Thrombocytopenic purpura
•
Decreased platelet production (e.g., myelofibrosis,
leukemia, myelodysplastic syndromes, chemotherapy,
radiotherapy, skeletal metastasis)
• Increased platelet destruction (e.g., immune
thrombocytopenic purpura)
• Platelet sequestration (e.g., splenomegaly)
Disorders of coagulation
•
Inherited (e.g., hemophilia A and B)
• Acquired (e.g., liver disease, vitamin K deficiency,
medication [warfarin, heparin])
Treatment
Common Initial Treatment
1.
Bleeding can usually be controlled by local measures:
• Pressure with moistened gauze
• Absorbable gelatin, absorbable collagen, microfibrillar
collagen or oxidized regenerated cellulose (to provide
a scaffold for platelets to adhere)
• Thrombin (to convert fibrinogen to fibrin)
• Epsilon-aminocaproic acid or tranexamic acid oral
rinses (to reduce fibrinolytic activity)
• Soft diet and avoiding factors that may provoke
bleeding, such as strenuous activities, traumatic
brushing, flossing, and rinsing
• If pre-existing dental models are available, a vacuum-
formed splint (+/− lined with thrombin powder) can
be fabricated and used to apply additional pressure
and protection
2.
Screening serologic studies should be performed.
• Complete blood count (CBC) with platelet count
• International normalized ratio (INR): measures the
factors of the extrinsic and common coagulation
pathways
• Partial thromboplastin time (PTT): measures the
factors of the intrinsic and common coagulation
pathways
• Thrombin time (TT): tests the ability of fibrinogen to
form an initial clot
• Platelet function analyzer (PFA-100) or Ivy bleeding
time (BT): screens for functional platelet disorders
3.
If the bleeding cannot be controlled, assessment with a
physician and systemic measures are necessary.
4.
Definitive medical management depends on the nature
of the underlying disorder. Principal agents for systemic
management include platelet infusion, fresh frozen plas-
ma, factor concentrates, cryoprecipitate, desmopressin
(DDAVP) and antifibrinolytic therapy.
2
. Inspect the visible skin and perform an intraoral
examination.
3.
Assess for other potential causes of oral hemorrhage:
• Mucocutaneous disorders (e.g., desquamative
gingivitis [erosive lichen planus, pemphigus vulgaris,
mucous membrane pemphigoid and erythema
multiforme])
• Necrotizing ulcerative gingivitis
• Drug-induced gingival hyperplasia
• Gingivitis of a local or endocrine (puberty, pregnancy)
cause
• Periodontitis
Hemorrhage that is evoked with minimal provocation or
spontaneously, especially if prolonged and difficult to control,
should alert the clinician to an underlying bleeding disorder.
Diagnosis
•
A hematologist will perform a focused history, physical
examination, and laboratory studies that may include
specific coagulation factor assays, mixing studies and
platelet aggregation tests.
•
If leukemia is suspected, diagnosis is confirmed by
peripheral blood smear and bone marrow biopsy for
cytology, immunophenotyping, and molecular/
cytogenetic studies.
Differential diagnosis
•
Local pathologies (see the Investigation section)
•
Systemic disorders:
Nonthrombocytopenic purpura
• Vascular disorders (e.g., scurvy, Ehlers-Danlos syndrome,
hereditary hemorrhagic telangiectasia)
• Disorders of platelet function (e.g., inherited disorders
[Bernard-Soulier syndrome, von Willebrand disease],
medications [ASA, NSAIDs], alcoholism, uremia)
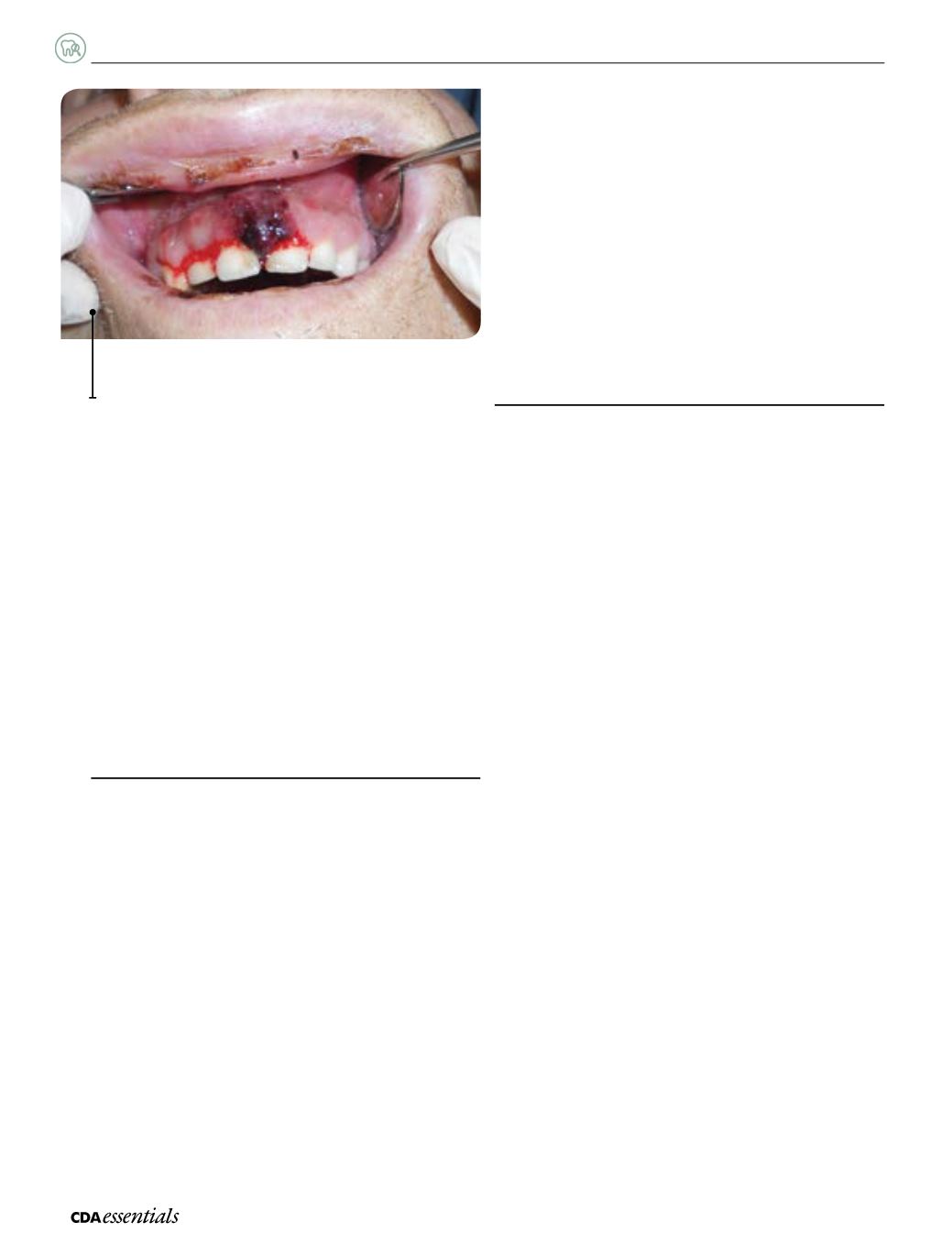
Fig. 1:
Leukemic gingivitis with prolonged gingival hemorrhage
following minor provocation.